Nurtuje Cię pytanie o długość życia osób z fenyloketonurią (PKU)? Rozumiem te obawy, ponieważ diagnoza choroby genetycznej zawsze budzi wiele pytań o przyszłość. Ten artykuł ma na celu rozwianie wszelkich niepokojów i dostarczenie rzetelnych, opartych na aktualnej wiedzy medycznej informacji. Dowiesz się, że dzięki nowoczesnym metodom leczenia, osoby z PKU mogą prowadzić długie, pełne i wartościowe życie, a diagnoza tej choroby nie oznacza skróconej egzystencji. Moje doświadczenie pokazuje, że świadomość i konsekwencja są tutaj kluczowe.
Fenyloketonuria: Długie i pełne życie jest możliwe
- Prawidłowo leczona fenyloketonuria (PKU) nie skraca długości życia.
- Kluczowe jest wczesne wykrycie choroby poprzez badania przesiewowe noworodków.
- Podstawą terapii jest restrykcyjna dieta niskofenyloalaninowa stosowana przez całe życie.
- Nieleczona PKU prowadzi do poważnych, nieodwracalnych uszkodzeń mózgu.
- Dzięki wczesnej interwencji pacjenci z PKU mogą osiągnąć normalny rozwój i prowadzić aktywne życie.
Fenyloketonuria a długość życia: Co mówi współczesna medycyna?
Główny niepokój rozwiany: jaka jest oczekiwana długość życia z PKU?
Wielu rodziców i pacjentów z fenyloketonurią (PKU) zadaje sobie pytanie o długość życia. Z mojego punktu widzenia, jako specjalisty, mogę z całą pewnością stwierdzić, że obecny stan wiedzy medycznej jasno wskazuje, iż prawidłowo leczona fenyloketonuria nie wpływa na skrócenie długości życia. To niezwykle ważna informacja, która powinna przynieść ulgę. Kluczowe jest wczesne wykrycie choroby oraz ścisłe przestrzeganie zaleceń terapeutycznych przez całe życie pacjenta. Diagnoza PKU nie jest wyrokiem, lecz początkiem drogi, na której konsekwencja w leczeniu pozwala na pełne i zdrowe życie. Według danych leki.pl, prawidłowo leczona fenyloketonuria nie wpływa na skrócenie długości życia, co potwierdza optymistyczne rokowania.
Historyczna perspektywa vs. dzisiejsze realia: jak zmieniły się rokowania?
Aby w pełni zrozumieć, jak daleko zaszła medycyna, warto spojrzeć na historyczną perspektywę. Przed wprowadzeniem badań przesiewowych noworodków i skutecznych metod leczenia, rokowania dla osób z PKU były niestety znacznie gorsze. Nieleczona choroba prowadziła do ciężkich, nieodwracalnych uszkodzeń mózgu, co skutkowało głęboką niepełnosprawnością intelektualną i ruchową. To były czasy, kiedy diagnoza PKU rzeczywiście wiązała się z tragicznymi konsekwencjami dla rozwoju i jakości życia.
Na szczęście, dzięki postępowi medycyny i wczesnej interwencji, dzisiejsze realia są zupełnie inne. Pacjenci z PKU, u których choroba została wykryta we wczesnym etapie i którzy konsekwentnie przestrzegają diety, mogą osiągnąć normalny rozwój intelektualny i prowadzić aktywne życie. Ta zmiana jest wręcz rewolucyjna i stanowi dowód na to, jak ogromny wpływ ma wczesna diagnoza i odpowiednie leczenie na jakość i długość życia osób z chorobami genetycznymi.
Wczesna diagnoza: klucz do zdrowia i rozwoju z PKU
Badania przesiewowe w Polsce – dlaczego pierwsze dni życia są decydujące?
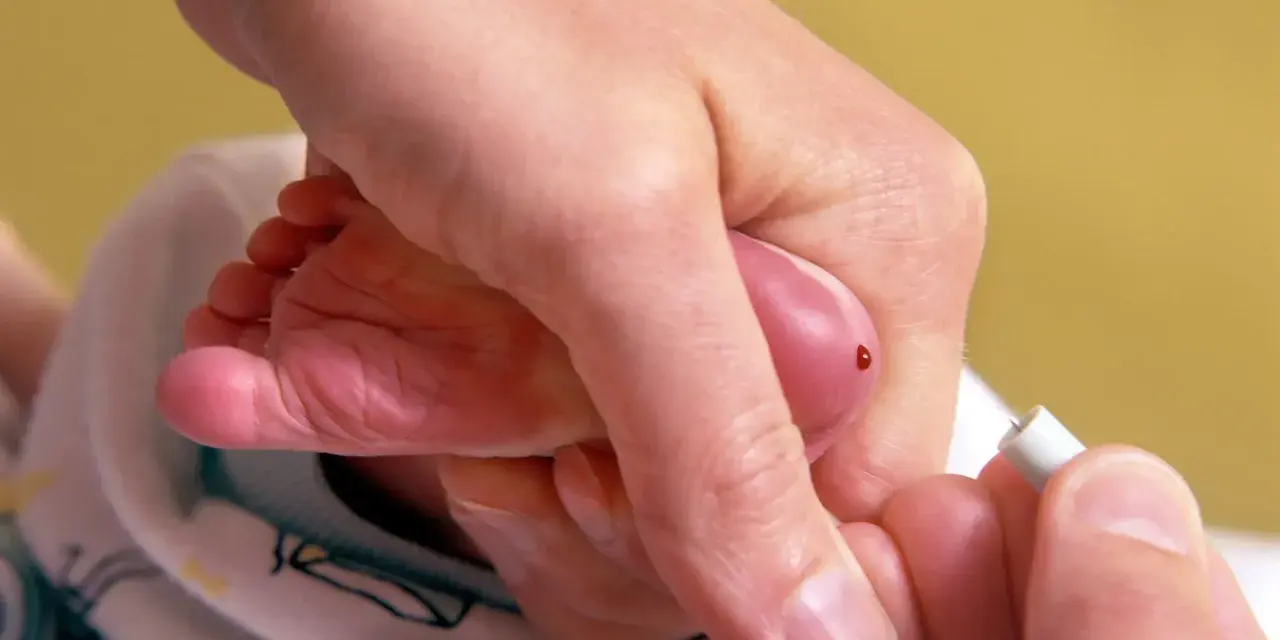
W Polsce mamy szczęście, że wszystkie noworodki są objęte obowiązkowym programem badań przesiewowych w kierunku fenyloketonurii. To niezwykle ważny element systemu opieki zdrowotnej, który ratuje tysiące dzieci przed nieodwracalnymi uszkodzeniami. Badanie to przeprowadza się zazwyczaj w 3. dobie życia dziecka. Polega ono na pobraniu kilku kropel krwi z pięty noworodka, a następnie oznaczeniu w niej stężenia fenyloalaniny – aminokwasu, którego nadmiar jest toksyczny dla mózgu. Dlaczego właśnie pierwsze dni są tak decydujące? Ponieważ wczesna diagnoza, najczęściej jeszcze przed ukończeniem pierwszego tygodnia życia, pozwala na wdrożenie leczenia dietetycznego przed upływem 14. doby. To krytyczne okno czasowe, które zapobiega nieodwracalnym uszkodzeniom ośrodkowego układu nerwowego i umożliwia dziecku prawidłowy rozwój.
Co oznacza pozytywny wynik testu? Kroki po diagnozie
Otrzymanie informacji o pozytywnym wyniku testu przesiewowego może być dla rodziców szokiem, ale ważne jest, aby pamiętać, że to dopiero pierwszy krok. Pozytywny wynik nie oznacza od razu ostatecznej diagnozy. W takiej sytuacji natychmiast podejmowane są kroki w celu potwierdzenia choroby. Dziecko jest kierowane do specjalistycznego ośrodka, gdzie przeprowadza się dodatkowe badania, aby jednoznacznie potwierdzić fenyloketonurię. Jeśli diagnoza zostanie potwierdzona, kluczowe jest jak najszybsze wdrożenie leczenia dietetycznego. Szybka reakcja i rozpoczęcie terapii pod stałą kontrolą specjalistów – lekarza genetyka, dietetyka, neurologa – jest absolutnie niezbędne dla zapewnienia dziecku szansy na normalny rozwój i pełne życie.
Dieta niskofenyloalaninowa: fundament terapii i gwarancja zdrowia
Na czym dokładnie polega "dieta PKU" i dlaczego jest niezbędna?
Dieta niskofenyloalaninowa, często nazywana po prostu "dietą PKU", jest fundamentem leczenia tej choroby. Jej głównym celem jest utrzymanie stężenia fenyloalaniny we krwi na bezpiecznym poziomie, aby zapobiec jej toksycznemu działaniu na mózg. Polega ona na eliminacji lub drastycznym ograniczeniu produktów wysokobiałkowych, które są bogate w fenyloalaninę. Do takich produktów należą między innymi mięso, ryby, nabiał (mleko, sery, jogurty), jaja, większość produktów zbożowych (pieczywo, makarony, ryż, kasze) oraz orzechy i rośliny strączkowe. Zamiast nich, pacjenci spożywają specjalistyczne preparaty białkozastępcze, które dostarczają niezbędnych aminokwasów (z wyjątkiem fenyloalaniny) oraz witamin i minerałów. Uzupełnieniem diety jest specjalna żywność niskobiałkowa, taka jak niskobiałkowy chleb, makarony czy ryż. Ta terapia musi być prowadzona przez całe życie pod stałą kontrolą lekarza i dietetyka, aby zapobiec gromadzeniu się fenyloalaniny i zapewnić prawidłowy rozwój.
Czy leczenie dietetyczne jest zobowiązaniem na całe życie?
Tak, leczenie dietetyczne w fenyloketonurii jest zobowiązaniem na całe życie. To jeden z najważniejszych aspektów, który zawsze podkreślam w rozmowach z pacjentami i ich rodzinami. Niestety, nie można go przerywać, nawet w dorosłym życiu. Przekonanie, że dorośli pacjenci mogą "wyrosnąć" z diety, jest błędne i może prowadzić do poważnych konsekwencji. Odstępstwa od diety, nawet sporadyczne, mogą skutkować wzrostem stężenia fenyloalaniny we krwi, co może prowadzić do pogorszenia funkcji poznawczych, problemów z koncentracją, pamięcią, a nawet zmian nastroju i zachowania. Konsekwentne przestrzeganie diety jest kluczowe dla utrzymania zdrowia neurologicznego, ogólnego dobrego samopoczucia i pełnej sprawności intelektualnej przez całe życie. To inwestycja w własne zdrowie i przyszłość.
Produkty dozwolone i zakazane: praktyczny przewodnik po żywieniu
Zrozumienie, co można jeść, a czego unikać, jest kluczowe w codziennym życiu z PKU. Poniżej przedstawiam praktyczny przewodnik:
| Produkty zakazane/ograniczone | Produkty dozwolone/specjalistyczne |
|---|---|
| Mięso (wołowina, drób, wieprzowina) | Specjalistyczne preparaty białkozastępcze |
| Ryby i owoce morza | Żywność niskobiałkowa (pieczywo, makarony, ryż, kasze) |
| Nabiał (mleko, sery, jogurty, twarogi) | Warzywa i owoce (w zależności od zawartości fenyloalaniny, pod kontrolą dietetyka) |
| Jaja | Tłuszcze roślinne (oleje, margaryny) |
| Rośliny strączkowe (fasola, groch, soczewica) | Cukier, miód, dżemy (w umiarkowanych ilościach) |
| Orzechy i nasiona | Napoje (woda, soki owocowe, herbaty) |
| Większość produktów zbożowych (tradycyjne pieczywo, makarony, ryż, kasze) | Specjalistyczne słodycze niskobiałkowe |
| Produkty zawierające aspartam |
Konsekwencje nieleczonej fenyloketonurii: ryzyko, którego można uniknąć
Toksyczny wpływ fenyloalaniny na mózg: ryzyko, którego można uniknąć
Kiedy stężenie fenyloalaniny w organizmie jest zbyt wysokie, aminokwas ten staje się toksyczny dla mózgu. Mechanizm jest złożony, ale w uproszczeniu polega na tym, że nadmiar fenyloalaniny zaburza transport innych ważnych aminokwasów do mózgu, co jest niezbędne do produkcji neuroprzekaźników – substancji chemicznych odpowiedzialnych za prawidłowe funkcjonowanie układu nerwowego. W konsekwencji dochodzi do uszkodzenia komórek nerwowych i zaburzeń w ich komunikacji. To ryzyko jest jednak całkowicie możliwe do uniknięcia. Wczesna diagnoza i konsekwentne leczenie dietetyczne pozwalają na utrzymanie fenyloalaniny na bezpiecznym poziomie, chroniąc mózg przed jej szkodliwym działaniem.
Obraz kliniczny nieleczonej PKU: od objawów neurologicznych po zaburzenia zachowania
Konsekwencje nieleczonej fenyloketonurii są niestety bardzo poważne i nieodwracalne. Ich znajomość jest kluczowa, aby zrozumieć wagę wczesnej interwencji:
- Ciężka, nieodwracalna niepełnosprawność intelektualna: Jest to najpoważniejszy i najbardziej znany skutek, prowadzący do znacznego opóźnienia w rozwoju poznawczym.
- Niepełnosprawność ruchowa: Mogą wystąpić problemy z koordynacją ruchową, spastyczność mięśni i trudności w chodzeniu.
- Padaczka: Ataki padaczkowe są częstym objawem u nieleczonych pacjentów.
- Zaburzenia zachowania: Nieleczona PKU może prowadzić do nadpobudliwości, agresji, autyzmu-podobnych zachowań oraz innych problemów psychicznych.
- Charakterystyczny zapach moczu i potu: Spowodowany nagromadzeniem metabolitów fenyloalaniny w organizmie.
- Jasna karnacja i włosy: Fenyloketonina wpływa na produkcję melaniny, co może prowadzić do jaśniejszej skóry i włosów.
Wszystkie te skutki są konsekwencją nieleczonej choroby i, co najważniejsze, można im skutecznie zapobiec dzięki wczesnej diagnozie i ścisłemu przestrzeganiu diety. To pokazuje, jak wielką wartość ma profilaktyka i konsekwentna terapia.
Jakość życia z fenyloketonurią: pełnia możliwości
Edukacja, praca, rodzina: realizacja życiowych planów z PKU
Jednym z najbardziej satysfakcjonujących aspektów mojej pracy jest obserwowanie, jak pacjenci z fenyloketonurią, dzięki prawidłowemu leczeniu, prowadzą pełne i szczęśliwe życie. Wczesna interwencja i konsekwentne przestrzeganie diety pozwalają im na osiągnięcie normalnego rozwoju intelektualnego. Oznacza to, że osoby z PKU mogą chodzić do szkoły, zdobywać wykształcenie, realizować swoje pasje, a następnie podejmować pracę zawodową. Są w stanie zakładać rodziny, wychowywać dzieci i spełniać się w różnych rolach społecznych, tak samo jak osoby bez PKU. Diagnoza fenyloketonurii nie jest przeszkodą w realizacji życiowych planów i marzeń, pod warunkiem odpowiedniego zarządzania chorobą.
Wyzwania dnia codziennego i rola wsparcia psychologicznego
Nie mogę jednak pominąć faktu, że życie z fenyloketonurią wiąże się z pewnymi wyzwaniami. Restrykcyjna dieta, konieczność ważenia produktów, planowania posiłków i unikania wielu potraw, zwłaszcza w sytuacjach społecznych, może być obciążająca. Może to prowadzić do poczucia izolacji, frustracji czy lęku, szczególnie u dzieci i młodzieży. Dlatego tak ważne jest wsparcie psychologiczne – zarówno dla pacjentów, jak i ich rodzin. Edukacja otoczenia (szkoły, przyjaciół, współpracowników) na temat PKU i specyfiki diety jest kluczowa dla budowania akceptacji i zrozumienia. Silne wsparcie ze strony rodziny, a także dostęp do grup wsparcia, pomaga pacjentom radzić sobie z wyzwaniami i budować odporność psychiczną, co przekłada się na lepszą jakość życia.
Przeczytaj również: Wzmocnij naczynia krwionośne - Koniec z pajączkami i obrzękami
Fenyloketonuria u dorosłych: na co zwrócić szczególną uwagę?
Fenyloketonuria w życiu dorosłym wymaga ciągłej uwagi i samodyscypliny. Konieczność kontynuowania diety i monitorowania stanu zdrowia pozostaje niezmienna. Dorośli pacjenci muszą regularnie wykonywać badania krwi w celu kontroli poziomu fenyloalaniny i dostosowywać dietę w razie potrzeby. Szczególnym aspektem jest planowanie ciąży u kobiet z PKU. W tym okresie utrzymanie ścisłej kontroli metabolicznej jest absolutnie kluczowe, aby zapobiec uszkodzeniom płodu (tzw. zespół fenyloketonurii matczynej). Kobiety z PKU planujące ciążę powinny skonsultować się ze swoim lekarzem i dietetykiem, aby zapewnić optymalne warunki dla zdrowia matki i dziecka. To pokazuje, że odpowiedzialność za dietę jest zobowiązaniem na całe życie, ale z odpowiednim wsparciem jest w pełni wykonalna.
Co przyniesie przyszłość? Nowe horyzonty w leczeniu fenyloketonurii
Medycyna nieustannie dąży do doskonalenia metod leczenia, a fenyloketonuria nie jest wyjątkiem. Patrząc w przyszłość, widzę wiele powodów do optymizmu. Trwają intensywne badania nad nowymi terapiami, które mogą jeszcze bardziej poprawić jakość życia pacjentów z PKU. Mówimy tu o innowacyjnych podejściach, takich jak terapie enzymatyczne, które mają na celu dostarczenie brakującego enzymu rozkładającego fenyloalaninę, czy terapie genowe, które dążą do korekty wadliwego genu odpowiedzialnego za chorobę. Choć te metody są jeszcze w fazie badań klinicznych lub wczesnego rozwoju, dają nadzieję na przyszłość, w której zarządzanie PKU będzie jeszcze łatwiejsze i mniej restrykcyjne. To pokazuje, że nauka nie stoi w miejscu, a pacjenci z fenyloketonurią mogą liczyć na coraz lepsze perspektywy.